IL-17是一种主要由Th17细胞产生的关键促炎症细胞因子,在多种自身免疫性疾病患者中高表达,包括银屑病、银屑病关节炎、类风湿关节炎和多发性硬化症等,并与这些疾病的发生发展密切相关。目前临床治疗主要采用抗体类疗法直接靶向IL-17或其受体IL-17R阻断其信号转导。然而,除银屑病等少数疾病外,该疗法在多种IL-17相关疾病的临床试验中因无效甚至加重疾病而宣告失败或被迫中止。这些与其发病机理相矛盾的现象原因未明,不仅极大地限制了抗IL-17疗法在相关疾病中的应用前景,也提示IL-17信号中可能存在未知的分子事件。
研究人员首先发现磷酸酶SHP2在多种自身免疫性疾病的病灶组织、相关动物模型和IL-17A刺激的细胞上高表达。令人意外的是,高表达SHP2后,无IL-17A刺激时细胞内IL-17R信号依然存在,但抗IL-17A单抗GR1501和Cosentyx对IL-17A信号的抑制作用则完全消失。另外,在小鼠胶原关节炎模型中,第二次免疫(第24天)即开始用抗IL-17A抗体治疗,可显著改善关节炎。然而,当临床评分达到3分(SHP2表达升高)以后(第31天)再开始给药,抗体的改善作用几乎消失。研究者接下来发现星形胶质细胞特异性诱导敲除SHP2小鼠EAE发病延迟,且疾病严重程度降低,脊髓组织中CCL2、CXCL1和CXCL2等显著减少,表明SHP2对IL-17信号的增强和维持至关重要。研究者还发现这种现象并非星形胶质细胞所特有,可能是表达IL-17R的不同类型组织细胞的共同特征。
随后研究者考察了SHP2是如何参与IL-17信号转导的。通过多种实验证实,SHP2可以和IL-17通路重要接头蛋白Act1相互作用,并且占据了TRAF5所结合的Act1的TRAF结构域,形成IL-17R-Act1-SHP2复合物,从而介导IL-17R信号的自激活。进一步发现,这种自激活依赖于SHP2的酶活,而SHP2发挥磷酸酶作用的底物是Act1,IL-17A刺激下Act1的酪氨酸磷酸化水平降低,SHP2 可以直接使Act1的Y548位发生去磷酸化,从而取代Act1-TRAF5复合物并激活IL-17R信号转导,促进相关疾病进展。这些结果表明,SHP2介导的Act1酪氨酸去磷酸化是IL-17R信号自激活的关键步骤,
研究人员随后证实,抗类风湿性关节炎药物艾拉莫德可以直接竞争结合Act1的TRAF结构域,分别阻断Act1与TRAF5和SHP2的相互作用,从而实现对基础IL-17信号和持续IL-17R信号转导的双重抑制。其在阻止疾病进展方面与抗IL-17抗体有着截然不同的表现,在上述抗IL-17抗体给药无效的时间段(第31天开始)给予艾拉莫德仍显著抑制小鼠胶原关节炎进展,这也提示了靶向自激活机制具有良好的应用前景,可为今后的新药研究奠定基础。
综上所述,该研究揭示了基础IL-17信号中一个过去未被报道过的“后门”机制——由IL-17A诱导SHP2高表达,随后形成IL-17R-Act1-SHP2复合物,介导IL-17R信号自激活。必须指出的是,这一后门机制系IL-17A本身主动设置,目的是加速和维持炎症的长期持续,并抵御相应的抗IL-17疗法。该研究首次回答了长期以来IL-17炎症为什么会走向慢性化的问题,解释了抗IL-17疗法对于相关疾病的临床试验为什么屡遭失败的原因,还展示了靶向该自激活机制的应用前景。类似这样的“后门”机制也可能存在于其他炎症信号通路中,可望为慢性炎症性疾病的治疗开辟新的途径。
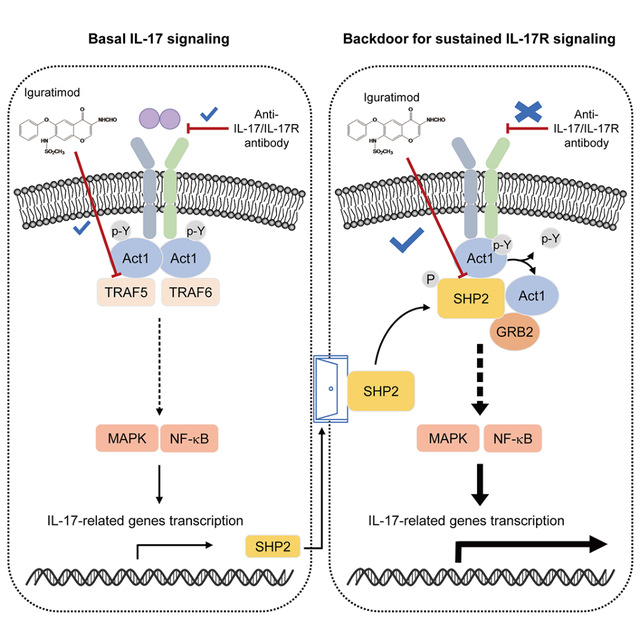
图基础IL-17信号及其“后门”IL-17R信号
该研究以“An autonomous activation of interleukin-17 receptor signal sustains inflammation and promotes disease progression”为题于2023年7月19日在线发表于免疫学顶级期刊Immunity。伟德BETVlCTOR1946徐强教授为该论文的主要通讯作者,伟德BETVlCTOR1946特任副研究员罗琼为第一作者和共同通讯作者。该研究受到国家自然科学基金重点项目等的资助。
原文链接:https://www.sciencedirect.com/science/article/pii/S1074761323002698。