The process of macrocyclization from linear precursors to cyclic structures can greatly alter the three-dimensional structure of molecules and thereby affect their biological function. Thioesterase (TE)-mediated macrocyclization is considered the most typical strategy for macrocyclization in polyketide and nonribosomal peptide biosynthesis. Additionally, P450, Rieske-type oxygenases, and radical enzymes can catalyze macrocyclization, such as in the construction of macrocycles in vancomycin, metacycloprodigiosin, and streptide. In addition, nature also employs some unconventional ways to construct macrocyclic scaffolds. For example, Diels-Alderase catalyzes the cyclization reaction to construct the macrocycle in pyrroindomycin, and the bifunctional oxidase LkcE catalyzes unique amide oxidation and Mannich reactions to construct the macrocyclic system in lankacidin. Therefore, understanding how nature generates macrocyclic molecules from acyclic precursors can provide new inspiration for the biological/chemical construction of functional macrocyclic molecules.

Alchivemycin A (AVM, 1) is a highly oxidized antimicrobial polyketide compound isolated from the endophytic plant Streptomyces sp. TP-A0867. It has a complex structure with a 17-membered N-heterocycle, a decalin ring, a rare 2H-tetrahydro-4,6-dioxo-1,2-diazine ring (TDO), an epoxy ring, and 16 chiral centers. In previous studies, Professor Guo Huiming's team from the School of Life Sciences at Nanjing University elucidated the secondary metabolism network involving six oxidoreductases during AVM biosynthesis, including unique intermolecular oxygen insertion into an amide bond (J. Am. Chem. Soc. 2021, 143, 4751−4757), but the process of its macrocyclization remained unknown. Recently, the team continued to analyze this macrocyclization process.
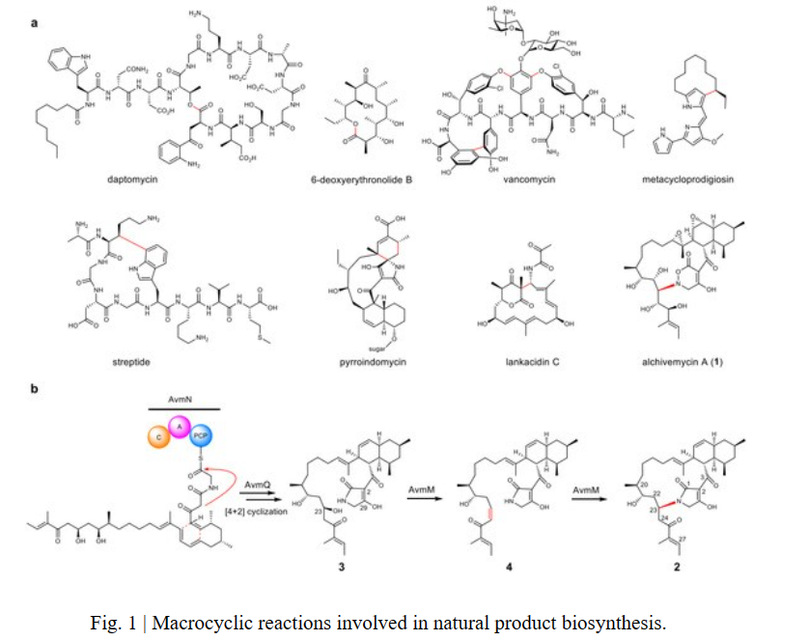
To elucidate its cyclization mechanism, the authors first knocked out the remaining uncharacterized genes in the gene cluster and found that avmM was associated with the formation of the 16-membered macrolactone in compound 2. Further in vitro enzyme catalysis showed that AvmM can rapidly cyclize compound 3 to form 2 without requiring any cofactors. The cyclization of compound 3 to 2 involves the departure of the hydroxyl group and the formation of a C-N bond, but the reaction result is inconsistent with an SN reaction. Deuterium labeling experiments found that the α-H of the carbonyl group at C25 was labeled with a D, indicating that β-elimination to form a double bond occurred first in the reaction, followed by a Michael addition to cyclize the molecule, although an intermediate dehydrated double bond was not captured in the reaction time course.
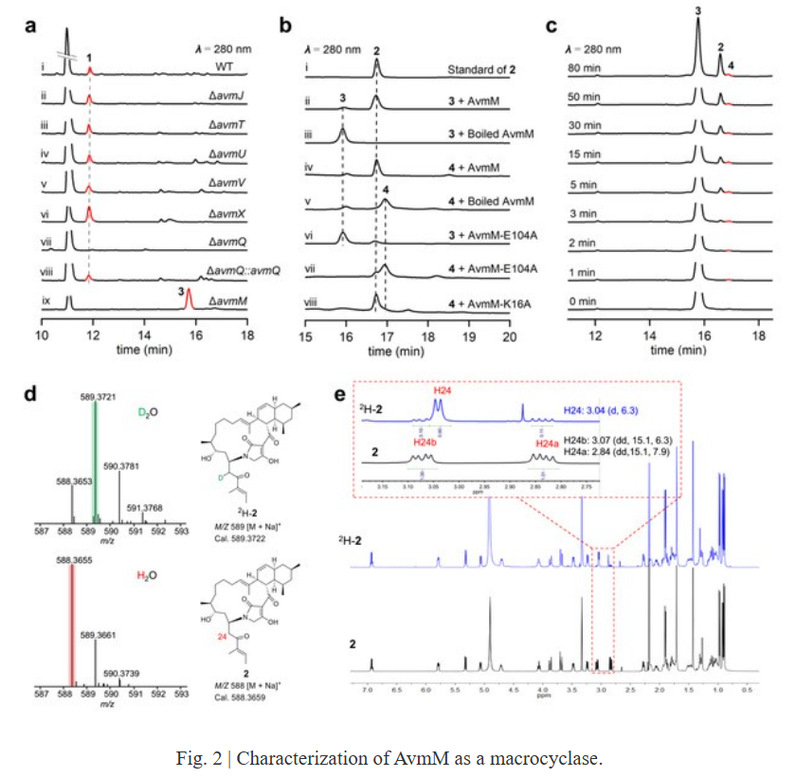
Subsequent bioinformatics analysis revealed that no protein similar to AvmM was found in the database, whether at the primary sequence or tertiary structure level. To further understand the catalytic process of AvmM, the phase was determined by introducing selenomethionine into the protein, and the crystal structures of AvmM (2.09 Å) and AvmM-2 (2.5 Å) complex were finally obtained. It was found that AvmM exists in the form of a homotrimer composed almost exclusively of beta folds.

Subsequently, combined with crystal structures, molecular docking and molecular dynamics simulations, point mutations were performed to verify the active site of the protein. Meanwhile, trace amounts of an unstable, speculated dehydrated intermediate were detected in the catalysis of AvmM-L182A, and eventually identified as dehydrated compound 4.
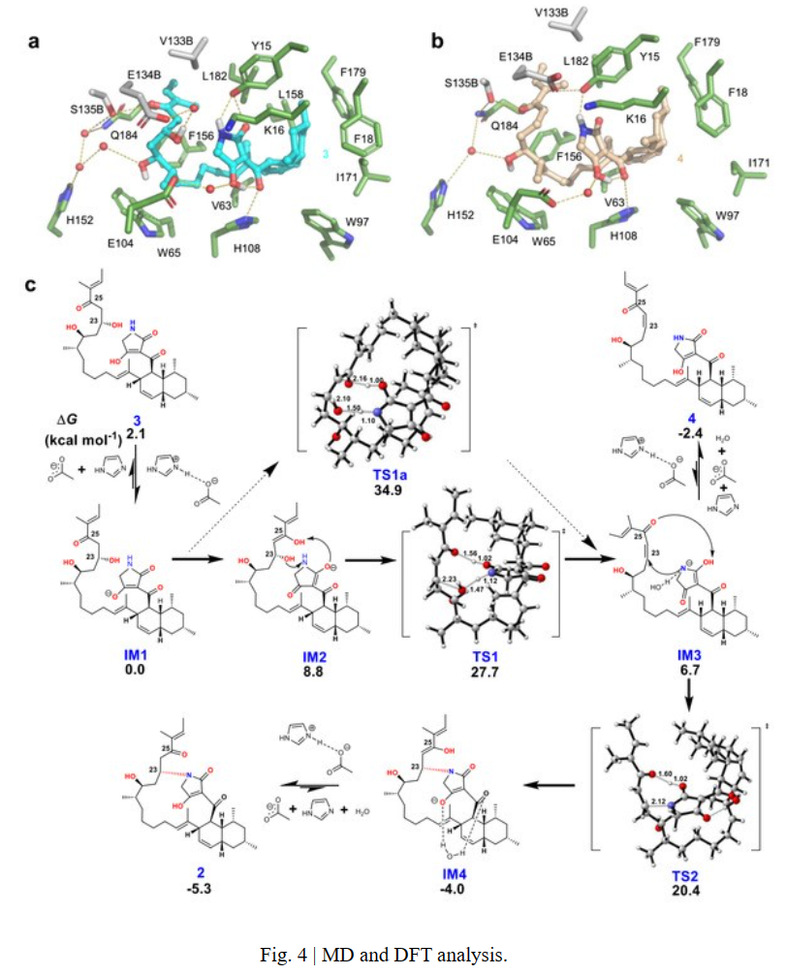
Unlike the classical PKS dehydration step that requires histidine to extract the α-H of the carbonyl group and provide a proton for the dehydration of β-OH, no amino acid residue within a 5 Å range of substrate 3 could act as a base to remove the α-H (H-24) of 3. To reveal its unique reaction mechanism, DFT calculations were further carried out. Based on all the above results, the catalytic mechanism of AvmM was analyzed: key residues E104 and H108 could initiate the dehydration reaction by proton transfer through the tetramer part, followed by N attack on β-C (C-23) for Michael addition, and the protonation of the enol intermediate resulted in the formation of cyclic product 2. At the same time, the formation of the 16-membered ring can significantly change the conformation of the flexible side chain in substrate 3, leading to the disappearance of hydrogen bonding interactions between the side chain and protein residues, facilitating the release of product 2 to complete the catalytic cycle.
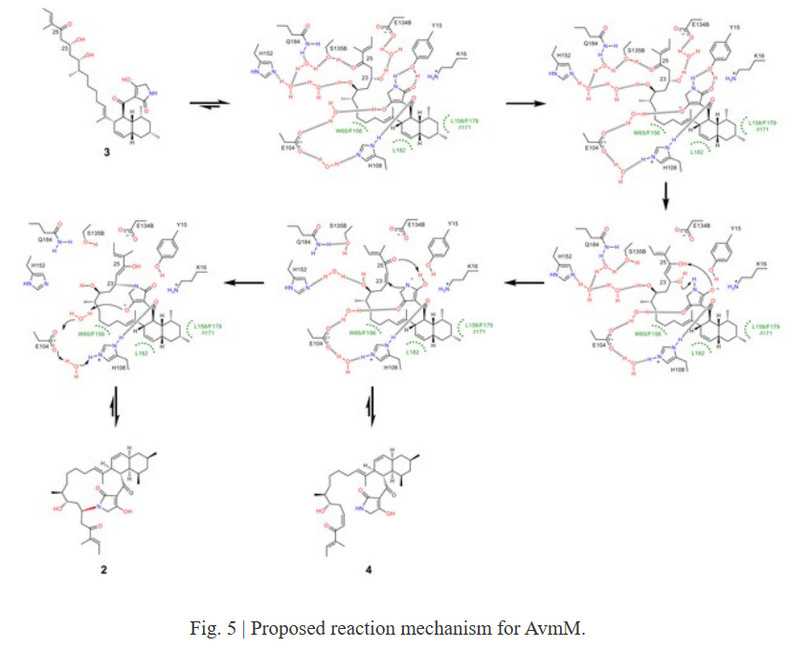
In summary, the authors detailed the mechanism by which AvmM catalyzes the construction of a 16-membered macrocycle through tandem β-elimination and Michael addition, revealing a new strategy for macrocyclization in natural product biosynthesis using in vivo gene knockout, in vitro biochemical experiments, X-ray crystallography, MD simulation, and DFT calculation. This study provides insights for enzymatic or chemical synthesis of functional macrocycles. The work was published in Nature Communications under the title AvmM catalyses macrocyclization through dehydration/michael-type addition in alchivemycin A biosynthesis.
Dr. Hong Jie Zhu and Associate Professor Bo Zhang from the School of Life Sciences, and Dr. Wan Qing Wei from the School of Chemistry and Chemical Engineering at Nanjing University are the joint first authors of the paper, while Professor Hui Ming Ge, Professor Ren Xiang Tan from the School of Life Sciences, and Professor Yong Liang from the School of Chemistry and Chemical Engineering are the corresponding authors. This research was supported by the National Science Fund for Distinguished Young Scholars and the National Key R&D Program.
Link to the original article: https://www.nature.com/articles/s41467-022-32088-4